Tag Archive for: MD Anderson cancer center
Understanding Myeloma Therapy Targets BCMA and GPRC5D
Understanding Myeloma Therapy Targets BCMA and GPRC5D from Patient Empowerment Network on Vimeo.
What are myeloma targets, and how do they impact the effectiveness of therapy? Dr. Krina Patel explains how treatments like bispecific antibodies and CAR T-cell therapy are using myeloma targets such as BCMA (B-cell maturation antigen) and GPRC5D (G protein-coupled receptor 5D) to kill myeloma cells.
Dr. Krina Patel is an Associate Professor in the Department of Lymphoma/Myeloma at The University of Texas MD Anderson Cancer Center in Houston, Texas. Dr. Patel is involved in research and cares for patients with multiple myeloma.
Related Resources:

Will CAR T-Cell Therapy Be Approved for Earlier Lines of Myeloma Treatment? |

|

How Can Myeloma Patients Access CAR T-Cell Therapy Clinical Trials? |
Transcript:
Katherine:
We know that the currently approved bispecific antibody therapies target BCMA and GPRC5D. What are these targets precisely and how do they impact the effectiveness of the treatment?
Dr. Krina Patel:
No, it’s a great question.
And, again, so BCMA we’ve had for a little bit longer.
We’ve known about it for a little bit longer, B cell maturation antigen, which definitely we’ve used as much as we can. So, we’ve had CAR Ts for it. We’ve had bispecifics for it. We’ve had antibody drug conjugates that we’ve attached to it.
So, it’s a really good target that is mostly just on myeloma cells and on very few other cells in the body, for the most part, which is why it makes such a great target. The side effects really should happen only specifically against the myeloma; so, less side effects in terms of toxicity. That’s not 100 percent the case.
BCMA is in some other tissues, like maybe the nerves, and that’s why maybe we see this toxicity sometimes, potentially in the GI system. Some patients can have it in other places. If you have myeloma in, let’s say, areas like the kidney. If you have a plasmacytoma, it can go to the kidney, things like that.
But again, for the most part, mostly on myeloma. And what’s really important about these targets is, once you get a treatment for it, what happens to that target. So, that’s a little bit different between these two targets. So, BCMA is a part of the proliferation of myeloma cells. So, it actually helps the myeloma cell survive. And so, the myeloma cells really want that BCMA on there. Now, for CAR T, for the most part, we don’t see people losing BCMA. We might see it go down in the myeloma cells that are left. For some patients, the expression can go down. But for the most part, we’ll see it come back up a few months later if the myeloma’s coming back.
The way that resistance happens with BCMA is that, when people are on bispecifics, the other treatment, we can sometimes see the BCMA get mutated. And then, maybe the other therapies we have won’t go after it any more.
So, again, it’s not common, but that’s sorta something we look at when we talk about sequencing therapy or which therapy should we use first. Then, GPRC5D’s a little different.
So, again, mostly just on myeloma cells. But here, we do know it’s on something called epithelial cells, which is skin, nails, tongue. And that’s why some of the side effects that we see, especially with the bispecific that’s a standard of care already, talquetamab, is skin and nail changes. So, people can get sloughing of their hands and nails; that can get disrupted. And then, taste. People can actually have some significant taste loss, to the point that they can have weight loss from it.
So, this is why that part is so important that if we have patients with these side effects, we need to hold the drug or decrease it; so, make sure we can turn those around. And then, the way GPRC5D is we think that it’s a little bit more likely that you can lose it once you get a treatment with GPRC5D that the myeloma can actually learn how to shed the antigen.
So, again, this really becomes important when we talk about combination and sequencing of all these different therapies we have and what’s the best way to do it so that patients can have the best response and the longest response.
How Is CAR T-Cell Therapy Research Advancing Myeloma Care?
How Is CAR T-Cell Therapy Research Advancing Myeloma Care? from Patient Empowerment Network on Vimeo.
What progress is being made in furthering advancing CAR T-cell therapy for myeloma? Dr. Krina Patel discusses the manufacturing process for CAR T-cells, research updates for manufacturing CAR T-cells faster, and the benefits of bridging therapy for some patients.
Dr. Krina Patel is an Associate Professor in the Department of Lymphoma/Myeloma at The University of Texas MD Anderson Cancer Center in Houston, Texas. Dr. Patel is involved in research and cares for patients with multiple myeloma.
Related Resources:
|
How Can Myeloma Patients Access CAR T-Cell Therapy Clinical Trials? |
Will CAR T-Cell Therapy Be Approved for Earlier Lines of Myeloma Treatment? |

|
Transcript:
Katherine:
Are there other advances in CAR T-cell therapy that patients should know about?
Dr. Krina Patel:
Yeah, so I think part of the issue right now is manufacturing and how long it takes for patients to get those cells. So, we use it to our advantage in the sense that earlier-line patients will have bridging therapy that we can give them while we’ve collected their cells and they’re being made; it takes are 4 to 6 weeks, or even eight weeks sometimes that we can give them a therapy that can knock their myeloma down before they get the CAR T.
And again, this is really important that we have options available. So, in fifth-line we don’t have very many options available. So, a lot of my patients, we really are just struggling to keep the myeloma controlled, try to bring it down before they get their CAR T. We’re hoping that that CAR T comes in any day.
When it goes earlier, I’m hopeful that now we’ll have options to actually bridge patients better because we’ll have more therapies they haven’t had. And the reason that bridging is so important is it really does decrease toxicity, some of the serious toxicity with see with CAR T; significantly decreases it.
And the efficacy. We see patients will do much better for longer if they have less myeloma going in than lot of myeloma going in. And so, again, I think because of that time, if we could get those cells earlier, that just makes it so much easier for all our patients to make sure that they’re able to get the cells. So, there’s quite a few different trials looking at fast CAR T production.
And so, there’s the PH383, I think. I can’t remember the number exactly. But this is one of the studies that was happening at Dana-Farber, and Dr. Sperling has presented couple time. The cells are made within just 24, 48 hours. And then, they actually go in and as they’re killing the myeloma, they grow.
So, they grow inside the body which is really, really, I think, a interesting way to develop CAR Ts for the future, make it more applicable and accessible. And then, there’s other companies in China. There’s the FasT CAR, which is a CD-19 plus BCMA, so two targets. But again, they can make their CAR Ts within a week.
And in the end, you have to still do quality checks for the FDA, which still take two weeks. So, it always will still be a few weeks, but still, the faster you can make those CAR’s, the more likely our patients are gonna be able to get it. And then, I think the combination studies. Again, there’s gonna be studies with different targets. So, there’s two CAR Ts, again, GPRC5D, that are going to be tested in the U.S.
A phase two study. And then, also another phase one study. And then, the phase two study, that GPRC5D CAR T is going to have combination studies coming out very, very soon. Actually, it’s already open in some places, and more places that are opening soon.
So, I think, yes, a lot’s going on again with new antigens and combinations.
How Can Myeloma Patients Access CAR T-Cell Therapy Clinical Trials?
How Can Myeloma Patients Access CAR T-Cell Therapy Clinical Trials? from Patient Empowerment Network on Vimeo.
How can patients learn more about joining myeloma CAR T-cell therapy clinical trials? Dr. Krina Patel shares advice for identify and accessing these trials, noting that seeking care with a myeloma specialist can be most helpful.
Dr. Krina Patel is an Associate Professor in the Department of Lymphoma/Myeloma at The University of Texas MD Anderson Cancer Center in Houston, Texas. Dr. Patel is involved in research and cares for patients with multiple myeloma.
Related Resources:
|
Will CAR T-Cell Therapy Be Approved for Earlier Lines of Myeloma Treatment? |
|

|
Transcript:
Katherine:
How can patients find and access clinical trials that are looking at CAR T-cell as an earlier line of therapy?
Dr. Krina Patel:
That’s a great question. So, I think going to any place that is myeloma specific. So, basically a big center that has doctors that are doing myeloma research, they will be able to definitely get you into places that have some of these trials. But clinicaltrials.gov is one other place. It’s really hard. I will tell you that, if I wasn’t a physician or in medicine, I don’t think I would learn, I would be able to navigate it very well.
And so, really either through your doctor and having them look this up for you, or going to patient groups. So, again, a lot of my patients are part of different patient groups where people will say, “Well, this is a trial that I was” or “This is a trial that my doctor told me about.” And then, asking. So, that’s the other big thing is constantly asking your doctor “What are my other options?” getting second opinions from myeloma experts, and then just paying attention to some of these resources that you have available. Right now, there are gonna be more clinical trials for earlier-line therapies and first-line with both cilta-cel and ide-cel.
There’s going to be clinical trials with new products: ddBCMA CAR T, that is likely gonna come out soon for earlier-line therapies. And so, there’s a lot happening, and so there might be different clinical trials in different places. But I think the fact that all this is going on at once is really important for our patients to know about.
Will CAR T-Cell Therapy Be Approved for Earlier Lines of Myeloma Treatment?
Will CAR T-Cell Therapy Be Approved for Earlier Lines of Myeloma Treatment? from Patient Empowerment Network on Vimeo.
Is there an opportunity for myeloma patients to gain access to CAR T-cell therapy sooner? Dr. Krina Patel discusses the results of clinical studies for CAR T-cell therapy and the potential for patients receiving the treatment earlier in their myeloma journey.
Dr. Krina Patel is an Associate Professor in the Department of Lymphoma/Myeloma at The University of Texas MD Anderson Cancer Center in Houston, Texas. Dr. Patel is involved in research and cares for patients with multiple myeloma.
Related Resources:
|
How Can Myeloma Patients Access CAR T-Cell Therapy Clinical Trials? |
|
|
Transcript:
Katherine:
Dr. Patel, current CAR T-cell therapy is FDA approved for patients who have had several lines of treatment.
But we know that there are a number of trials that are exploring this treatment in earlier lines. So, what is the progress on these trials?
Dr. Krina Patel:
Yeah. So, I think we have two major ones that have already been done and we’ve heard the results from. So, CARTITUDE-4 was for cilta-cel in second to fourth-line; so, patients who have relapsed once, all the way up to three times.
And then, KarMMa-3, which is ide-cel, which was one line later. So, that was third-line to fifth-lines; so, relapsed twice to four times. So, little bit different patient populations in the two trials. The trials were different in that patients had different therapies before too.
But both were positive studies which was what was really exciting. So, in CARTITUDE-4 patients were randomized, meaning they got either the CAR T or they got a standard of care option. And the CAR T won by a lot. This was, we call, hazard ratios.
But basically, the amount of different of patients surviving when they got CAR T without myeloma versus the standard of care was one of the biggest differences we’ve ever seen in a clinical trial for multiple myeloma. So, it’s –
Katherine:
Wow.
Dr. Krina Patel:
– something really pretty amazing. And then, KarMMa-3, that trial, same thing. There’s a huge difference in the patients who got CAR T versus the standard of care. The standard of care options were different in the two trials for the most part. So, again, different patient populations and different standard of care options, but the other big thing that the KarMMa-3 study did was they allowed for patients who are on the standard of care that, once they were relapsing, they could get the CAR T.
And so, because we have this crossover the big controversial thing that came up was, “Well, patients aren’t necessarily living longer by getting CAR T earlier. As long as they get CAR T they do really well.” And so, that is why there was a big meeting with the FDA what we call the ODAC meeting.
So, they had both companies present their trials to the FDA and to this advisory board that they had called ODAC, and thankfully it was positive. So, both studies were positive in terms of the advisory board saying that they agreed these should be moved up forward.
So, now we’re just waiting and hoping the FDA approves them so that we can actually give it to patients. I think the biggest reason is access. So, we know that when patients are fifth-line, which is when it’s approved now, not everybody makes it to fifth-line. It’s really hard to get through all these therapies and then still be healthy enough to do this versus if it’s approved in second and third-line, that just means so many people can actually get these therapies and available to them.
And the other big thing is the quality-of-life piece for CAR T. It’s been such a big difference when patients get a break from therapy for a year or two years or longer compared to being on continuous therapy. And so, both studies have had quality of life studies come out as well showing that difference between the standard of care versus the CAR T.
Advances in Managing CAR T-Cell Therapy Side Effects
Advances in Managing CAR T-Cell Therapy Side Effects from Patient Empowerment Network on Vimeo.
What progress is being made in treating the side effects of CAR T-cell therapy? Myeloma expert Dr. Krina Patel discusses the research and advances being made in understanding and managing the common issues associated with this treatment.
Dr. Krina Patel is an Associate Professor in the Department of Lymphoma/Myeloma at The University of Texas MD Anderson Cancer Center in Houston, Texas. Dr. Patel is involved in research and cares for patients with multiple myeloma.
See More From Thrive CAR T-Cell Therapy
Related Resources:

|
Recovering From CAR T-Cell Therapy | What Can Myeloma Patients Expect? |

What Side Effects Are Possible Following CAR T-Cell Therapy? |
Transcript:
Katherine Banwell:
Dr. Patel, what about managing the side effects of CAR T? Is that improving?
Dr. Krina Patel:
It is. I think just the fact that we are now seeing less of the neurotoxicity that we were seeing in the initial patients that were getting CAR T, and the serious neurotoxicity like the Parkinsonianism or patients couldn’t walk or hold things because of the tremors they were having that were so significant.
And then something kinda like Guillain-Barré where people are getting ascending paralysis. And so, again, those are very serious things that we don’t want any of our patients to get. And again, most of it was with cilta-cel and their CARTITUDE-1 study. There is a black-box warning with ide-cel too that that can happen.
We just don’t see it at the same rate. And again, when patients got better bridging going into CAR T, that rate dropped from 6% to 0.5%. So, it really is a huge difference by just giving better bridging and having myeloma controlled better going in.
We are seeing some other side effects that we didn’t necessarily see on the original clinical trials, like facial palsies. These are things where patients can talk very well anymore or their side of their face is drooping. And those are reversible thankfully, for the most part with steroids, things like that. So, we watch for that. There’s some other side effects.
Again, these are immune cells going into your body, so anything can happen. So, we definitely keep a close eye on all of our patients. But for the most part, especially things like infections and this neurotoxicity, we’ve learned that again the less myeloma going in, the better patients do and the faster they recover.
So, for the most part, patients now are doing much, much better than when patients were originally on those trials where we didn’t know what was gonna happen. And I think the infection piece is such a big deal; that our patients definitely need supportive care.
So, even though this is a one-and-done where you get your chemo, you get your CAR T’s and then you’re not on any myeloma therapy, you are on supportive therapies; so, things like IVIG for your immune system, things to protect you against something called PJP pneumonia for at least six months if not longer.
You’re still on anti-shingles medication. So, it’s not that you stop all medicines, but it’s you stop the myeloma medicines. And then, at least for the first six months, maybe even up to a year we have to make sure you don’t get any major infections.
Accessing Quality Myeloma Care | Advice for Overcoming Obstacles
Accessing Quality Myeloma Care | Advice for Overcoming Obstacles from Patient Empowerment Network on Vimeo.
How can you access the myeloma care that is best for YOU? Myeloma specialist Dr. Krina Patel shares advice for patients, including the importance of a second opinion and key questions to ask your doctor regarding your disease and treatment plan.
Dr. Krina Patel is an Associate Professor in the Department of Lymphoma/Myeloma at The University of Texas MD Anderson Cancer Center in Houston, Texas. Dr. Patel is involved in research and cares for patients with multiple myeloma. Learn more about Dr. Krina Patel.
Related Resources:



Transcript:
Katherine:
What hurdles to patients face when accessing quality overall myeloma care and what can be done to get over these obstacles?
Dr. Krina Patel:
I talk about this a lot.
So, again I think the biggest problem for me is that because myeloma care changes so fast, which is a good thing that we have all these options and we have so many new therapies, it’s really hard for people who don’t do just myeloma to keep up. I don’t think I would be able to. I don’t do breast cancer. I don’t do other cancers, so when I take my boards every 10 years, I have to learn a lot to take those.
So, it’s just a part of the system that this the problem. So, I think if you’re seeing a local oncologist that sees five myeloma patients a year, they’re gonna be stuck on what was the treatment when they did it last time for that last patient, which again might be very different now because things change so fast.
And so, again, you want to get to a doctor quickly, and I understand that. When people hear “cancer,” they’re like “I gotta get treatment. I gotta go fast.” But part of it is, if you need treatment quickly to get to your doctor. But then, try to make a second-opinion appointment done, even virtually because we can do that now after COVID; we have so many more options for that.
And get that second opinion just to say “Is this the right therapy for me? Going forward, what should I do?” So, patients, “Should I get a stem cell transplant?” if you’re newly diagnosed or not. “What kinda maintenance should I be doing? Do I have high-risk disease or not? What are the nuances of my myeloma versus everybody else that we need to be careful about? Should we dose reduce?” There’s a lot of those types of hurdles. Patients, if they have kidney failure form their myeloma, we should be decreasing the dose of some of the medications; those types of things that really we can help with to make sure those outcomes are in the best.
And that first treatment really does matter so that we can reverse as much as possible, for patients who have kidney involvement versus bone involvement, to decrease the pain really quickly. Do we need to get our radiation doctors involved to get radiation to help make sure you don’t get a fracture from a potential bone lesion. So, I think, again, I understand the urgency of seeing somebody, of getting diagnosed, and starting therapy.
But quickly get to a second opinion so that they can help. And then, again, some of these patient advocacy groups are amazing for myeloma. And I think there’s just so much information there that you don’t want to get overwhelmed, but at the same time you want to start going a little bit at a time at those things so that you can learn more about what you need to be asking and doing.
Key Advice for Myeloma Patients | Questions to Ask About a Care Plan
Key Advice for Myeloma Patients | Questions to Ask About a Care Plan from Patient Empowerment Network on Vimeo.
How can newly diagnosed myeloma patients be proactive in their care? Dr. Krina Patel shares key advice for patients, including the importance of making notes before office visits and the role that a care partner can play in overall support.
Dr. Krina Patel is an Associate Professor in the Department of Lymphoma/Myeloma at The University of Texas MD Anderson Cancer Center in Houston, Texas. Dr. Patel is involved in research and cares for patients with multiple myeloma. Learn more about Dr. Krina Patel.
Related Resources:

Transcript:
Katherine:
For newly diagnosed patients, what key advice would you share with them? And are there specific questions they should be asking their doctor about their care plan?
Dr. Krina Patel:
Yeah. So, I know it’s hardest for newly diagnosed patients. Most people have not even heard what multiple myeloma is. They’re learning how to spell it correctly and making sure it’s not melanoma. And this is a conversation I have with so many of my new patients that I think it’s really hard your visit, and maybe even your second visit, to ask all the right questions. So, really, coming home and every time you’re on a treatment or you’re talking about a treatment and you have a question, write it down because I know it’s really hard when we’re only there for 15, 30 minutes to talk to you.
For us, we have MyChart, so my patients will send questions as they think of them through that. And I think that’s really important. Sometimes it’s hard to know what questions to ask when you have no idea what’s about to happen, and that’s okay. But I think as you’re going through therapy, really making sure that you ask about alternative therapies that might be available and why someone is picking one versus the other, making sure you know what supportive medications you really need.
And I will say that, with myeloma, a lot of our treatments are patient-friendly but they do cause side effects and infections, so, we have a lot of supportive medications we use; so, again, anti-shingles, potentially if you could get a blood clot, we have you on some type of blood thinner.
We have people on against steroids because of all of our initial therapies have steroids. We wanna make sure you don’t get ulcers in your stomach, so we have patients on proton pump inhibitors. There’s a lot of things we do to again decrease that toxicity. So, that’s important.
And then, I think the next part is when you’re on treatment, whatever symptoms you’re having keep a log of that. Some things are, okay, maybe it’s just a little bit here and there, that you’re feeling fatigued but then you’re better. But there are certain things that cause a lot of side effects that my patients sometimes don’t tell me about. So, the steroids can cause major insomnia for some of my patients where they don’t sleep for three days, and that’s not okay. We can decrease those.
So, there are ways to manipulate the treatments as we’re going through to make sure that not only are you having a great response but that you’re not having major side effects that are actually gonna hurt your health down the road. So, really important to discuss those things that you’re having as you’re going through.
Katherine:
There’s also the importance of a care partner in your life –
Dr. Krina Patel:
Yes.
Katherine:
– right?
Dr. Krina Patel:
I agree. So, I joke with my patients but it’s real; there’s actually a study that shows that men with three and a half women in their lives do much better in healthcare in general than those who don’t. So, I’m like “Go get more women in your life” –
Katherine:
I love that.
Dr. Krina Patel:
– or just caregivers in general.
Men are great caregivers too, but really having someone there that can listen for you and write down those things because it is overwhelming. And when you’re on treatment there are a lot of times when you just can’t pay attention. You can’t focus. You can’t listen to everything. And so, the more people that are there, they’ll pick up other things.
So, a lot my patients will even have their loved ones on their phone with them, even if they can’t be there in person so that they can record. And a lot of my patients will record things and they’ll ask me; so, definitely as whoever you’re talking to if it’s okay to record. But most of us will say “Yes, it’s completely fine” so that you can listen to it again when you go home.
What Is the Role of Bispecific Antibody Therapies in Future Myeloma Care?
What Is the Role of Bispecific Antibody Therapies in Future Myeloma Care? from Patient Empowerment Network on Vimeo.
How does bispecific antibody therapy impact the outlook for myeloma care and treatment? Dr. Krina Patel discusses how this treatment, and CAR T-cell therapy, are revolutionizing myeloma care.
Dr. Krina Patel is an Associate Professor in the Department of Lymphoma/Myeloma at The University of Texas MD Anderson Cancer Center in Houston, Texas. Dr. Patel is involved in research and cares for patients with multiple myeloma. Learn more about Dr. Krina Patel.
Related Resources:
Transcript:
Katherine:
What role do you foresee bispecific antibody therapies playing the future of myeloma care?
Dr. Krina Patel:
So, I think bispecifics are phenomenal. I’m a CAR T girl, but in terms of access, I will say, that more of our patients around the world are going to have access to bispecifics.
And it’s off the shelf, so you don’t have to worry about taking cells out, making it, waiting and hoping to get those cells back. So, many more patients are going to be able to get it. And I think ideally if everybody could get a CAR T, my goal would be a CAR T first and then a bispecific until we can cure myeloma. Unfortunately, for the most part right now with these therapies, as single agents we haven’t seen that the majority of patients are cured.
So, my goal is to make sure that I have all the treatment options possible to keep patients doing well for as long as possible. And so, again, ideally CAR T, then maybe a bispecific because of the way those resistance mechanisms happen. But if we don’t have the availability of CAR T for everybody or you’re not eligible, I do think bispecifics are a great therapy. And I have again patients who are frail, who are older that we’ve been able to give bispecifics to and they’ve had amazing responses. And I think right now they’re single agents. But I do think that as we get these trials with combinations approved, we’ll see a lot more increase in use of those.
Again, the side effects are still something we’re learning about. So, bispecifics with BCMA, infections are a really big risk, even more than CAR T.
So, it’s really, really important that, if anyone has fevers or they don’t feel well, they see their doctor right way and make sure it’s not a strange infection that we don’t usually see that needs to be treated versus even a regular pneumonia that can be pretty dangerous when your immune system’s down.
So, that’s important. And then, the GPRC5D, as I said, it’s the taste and the weight loss and things like on skin that we really wanna make sure we do as much supportive care for that as possible.
What’s Next in Myeloma Research and Treatment?
What’s Next in Myeloma Research and Treatment? from Patient Empowerment Network on Vimeo.
What are the next generation myeloma therapies? Dr. Krina Patel shares an update on new agents, such as CelMoDs, and discusses how combination treatment and sequencing of therapy will evolve in the future of myeloma care.
Dr. Krina Patel is an Associate Professor in the Department of Lymphoma/Myeloma at The University of Texas MD Anderson Cancer Center in Houston, Texas. Dr. Patel is involved in research and cares for patients with multiple myeloma. Learn more about Dr. Patel.
Related Resources:

|
Evolving Myeloma Treatment Options | Bispecific Antibody Therapy |
|
Transcript:
Katherine:
Dr. Patel, for the last few years advances in myeloma treatment have been focused on the cellular therapies like CAR T. Can you share what’s next in myeloma research and treatment?
Dr. Krina Patel:
Yes, I think it’s been really exciting. The last 10, 15 years, really, we’ve just catapulted in the whole world of immunotherapies; so, from monoclonal antibodies to even IMids and CelMoDs and things that we’ll talk about a little bit now, and cell therapy as well as just other ways of engaging T-cells with the bispecific therapies too. So, I think what’s really exciting, that we have not just new mechanism of action that’s coming down the road but new targets.
So, again, coming back to really the big stuff like immunotherapy that I really like a lot and what I really am excited about, we have different ways of using the immune cells to help fight myeloma.
And so, things like IMids, lenalidomide, and pomalidomide that are older drugs that we’ve had since 2006, but really there’s newer ones called CelMoDs that are coming out that are being evaluated in clinical trials. One is called iberdomide. Another is called mezigdomide.
So, we’re really excited about this really in combination therapy, just like the prior iMids were used. And what it really does is it improves your immune system; it activates it to a point where things like monoclonal antibodies, such as daratumumab or isatuximab or elotuzumab, can work better in synergy.
But even new trials with some of our CAR Ts that we already have available, the BCMA therapies, combining it with these to see if we can make those T-cells work better.
So, once you get the CAR T-cell infusion, can we give some of these therapies now to improve how long it lasts, how well they work. And the same thing with the bispecifics.
These are therapies that are using the T-cells that are already in your body. Can we combine it with some of these of other immune therapies that will help the T-cells already there get activated, and then the bispecific takes them to the myeloma to really get treated. And I think those combination studies that are coming down are really, really exciting. And then, I think the new antigens, as I mentioned, not just BCMA therapy but we have GPRC5D and we have something called FcRH5.
And to my patients, I say it’s alphabet and number soup basically but they’re really targets for myeloma that we’re finding. It’s pretty amazing, considering that we didn’t have a target for the longest time, like lymphoma when they had their CD19 and we were jealous. And now we’re finding all these targets and now we’re figuring out how do we combine different mechanism of action for different targets so that we can hopefully kill every last myeloma cell.
Dr. Nizar Tannir: Why Is It Important for You to Empower Patients?
Dr. Nizar Tannir: Why Is It Important for You to Empower Patients? from Patient Empowerment Network on Vimeo.
Renal medullary carcinoma (RMC) can be a devastating cancer, but healthcare providers can help make a substantial impact. RMC expert Dr. Nizar Tannir from MD Anderson Cancer Center shares how he creates a positive, healing relationship with patients and what he views as the future of RMC patient care.
See More from Empowering Providers to Empower Patients (EPEP)
Related Resources:

|

|

Dr. Ana Maria Lopez Why Is It Important for You to Empower Patients |
Transcript:
Dr. Tannir:
It all starts with listening to the patient and taking their history. And their symptoms very seriously not to dismiss what they tell you. That’s the first step patients who seek our help, they come to the provider, they come to the physician to help them seeking healing. I think it’s fundamental to listen, listen carefully take every symptom they report to us seriously. And when we think of a diagnosis of RMC as physician first, because the patient comes first before anything else. We provide them with all the information we have, we may not have all the information about their diagnosis or about the future.
When they ask us what will the future look like? What should they expect? We may not be able to answer that the question, but we can still provide them with help and take their question seriously and say, “I don’t know the answer to your question, I don’t know what the future will bring, but I’m going to tell you, I will not leave any stone unturned until I get to the bottom of it. And until I am able to find an answer to your question. “Then second, for young investigators who aspire to have a career in medicine, in medical research, in scientific research RMC is the most fulfilling field that you can make an impact on humanity.
For all of you who are ambitious, aspirational, hardworking, well-trained, smart, want to make a difference in the world and help humanity, RMC will provide you with a golden opportunity to make that difference, because patients with RMC are young, are all active before they come to us. Sick, debilitated, devastated with devastated family members. It is an aggressive disease that if not treated aggressively, urgently, unfortunately, patients may not make it. So it behooves us to provide them with the best care and research will give us the opportunity to, in the future, hopefully cure this disease once and for all. And what’s more rewarding for a career than seeing young patients achieve a cure from a devastating cancer that may, unfortunately, take their life away in few months or a year or two, if they can live for many many years to their fullest potential as a normal human being, to live to the final aging and give the society back, its citizens to be productive in society.
Give those young individuals the chance to go back to work or back to college. Maybe start a family, get married and have a family and have children. What is more rewarding than this? I think empowering yourself. Empower yourself with that golden opportunity. Empower yourself with that career that can help you make a difference in the world so that the world will not be deprived of young people like Herman Connor who could have not had that opportunity but now is alive and well and a productive member of society and a citizen 11 years after diagnosis. So imagine what you could do to help another patient like Herman and give that patient the opportunity to be cured.
A Renowned Expert Weighs in on the Future of Renal Medullary Carcinoma
A Renowned Expert Weighs in on the Future of Renal Medullary Carcinoma from Patient Empowerment Network on Vimeo.
What does the future of renal medullary carcinoma (RMC) care look like? Expert Dr. Nizar Tannir shares his perspective on the progression of RMC care, issues that still need advocacy, and his hopes for the future.
Dr. Nizar Tannir is a Professor in the Department of Genitourinary Medical Oncology, Division of Cancer Medicine at The University of Texas MD Anderson Cancer Center.
[ACT]IVATION TIP
“So, my hope and my plea is for all of us to work together to realize this dream that I hope I will see before I die and before I retire, that RMC deserves the support, not only RMC, but all aggressive, rare tumors require and deserve the support from funding and equal healthcare access.”
Download Guide | Descargar Guía
Related Resources:

|

How Do You Explain RMC to Newly Diagnosed Patients and Families? |
Why Renal Medullary Carcinoma Clinical Trial Participation Is Pivotal |
Transcript:
Cora:
Dr. Tannir, you’ve dedicated your life to patients like my brother Herman, and we are so grateful for your genius and that of your colleagues, what is your hope for the future of RMC?
Dr. Tannir:
Cora, it’s been a privilege, an honor to have been with Herman and you on his journey from April 2012 and today, April, we are in April 2023, 11 years. He came with stage IV, and it took several months trying to get to a place that will take his symptoms and his diagnosis, seriously, get to the bottom of it and initiate therapy urgently. I am so happy for the outcome that Herman has achieved with your support, I could not think of how he could have done it without you on his side. You have dedicated, in fact, I know you have three sons, you’ve raised three wonderful young men, but you had Herman always.
So my hope is Herman’s success story. Herman triumph over RMC, will not be just one singular story that we talk about, but that story, I hope will be the common story we will tell about other patients with RMC. It will require research. It will require funding for the research, it’ll require raising awareness. It’ll require working with our policy makers, our congress, members of Congress and administration to open the gates, remove the barriers for patients with RMC, who are in this country, who are U.S. citizens, to allow them the care that members of Congress get that I get and I can get at MD Anderson any time. I think it is important, it’s not just important to have the tools, the scientific tools, the research tools, but also if those tools, if those clinical trials are not made available to people with RMC who need it in every city and town in the United States, well, how good our tools are if they can be only offered to only a few.
So, my hope and my plea is for all of us to work together to realize this dream that I hope I will see before I die and before I retire, that RMC deserves the support, not only RMC, but all aggressive, rare tumors require and deserve the support from funding and equal healthcare access. We really need to remove those disparity barriers. But from the scientific research and medical standpoint, I really see that we have seen a sea of change over the past 10 years and more so over the past five, six years. We are confident that we can, we can with a red pen mark, cross that RMC I hope in the next few years.
I know we can do it, there has been, So much excitement about RMC, whereas 10 years ago when Herman was diagnosed, not too many people were excited or interested or knew about RMC and what to do, now many, I can say many investigators in the U.S. and in Europe and other places are interested. They’re doing research. They’re applying for grants on RMC. So I am optimistic, I am glad. I look back and I see that all that hard work, and I think Herman is a beacon, a bright light in this dark field that has been dark field RMC, that light that has shone it was shining, and you’re helping there to lead to improvement. I can tell you, Cora, that patients with RMC now have a much better chance of surviving many years compared to years ago when they only had a few months to live. Now people are living two, three, four, five, and seven years, and 10 years and longer. So progress has been made, but there is more…more progress to achieve towards the cure.
I am confident that we will conquer RMC in the future. It takes a village. It takes the world to do it, and I am happy to see that we have made that progress. So thank you for all the support you provided to all those patients. Not just your brother, but all these people who contacted you to get guidance and wisdom from you and for sending many of those my way and to MD Anderson. So it’s my privilege and honor to have been at MD Anderson in this field to be able to have helped your brother and help other patients with RMC together, we will do it, we will conquer RMC.
Share Your Feedback:
Create your own user feedback survey
Advice for Newly Diagnosed Renal Medullary Carcinoma Patients
Advice for Newly Diagnosed Renal Medullary Carcinoma Patients from Patient Empowerment Network on Vimeo.
What renal medullary carcinoma (RMC) advice should patients know about? Expert Dr. Nizar Tannir shares advice about seeking RMC information and ways to help maintain health and things for RMC patients to avoid.
Dr. Nizar Tannir is a Professor in the Department of Genitourinary Medical Oncology, Division of Cancer Medicine at The University of Texas MD Anderson Cancer Center.
[ACT]IVATION TIP
“…get the facts, get the right information, right facts, medical facts from reliable sources…get the rest they need, they should avoid strenuous, extreme intense exercise. They should eat healthy food well-balanced food, stay well-hydrated, seek the care at the best place they can have access to, and keep the hope alive…do not panic. There is hope.”
Download Guide | Descargar Guía
Related Resources:

|
How Do You Explain RMC to Newly Diagnosed Patients and Families? |

Renal Medullary Carcinoma Treatment Options for Newly Diagnosed Patients |
Transcript:
Cora:
Dr. Tannir, what do you advise your newly diagnosed RMC patients and their care partners to not do?
Dr. Tannir:
The main message I have for patients with RMC and their caregivers partners is to not panic and important my advice is to tell them to get the facts, get the right information, right facts, medical facts from reliable sources, because there is a lot of stuff on social media that some of it may not be accurate. And so in terms of what they should not do other than they should not panic I think they should support their loved one in their journey as you supported Herman. They should get the rest they need, they should avoid strenuous, extreme intense exercise. They should eat healthy food well-balanced food, stay well-hydrated, seek the care at the best place they can have access to, and keep the hope alive. That’s my activation tip. Do not panic, do not panic, there is hope.
Share Your Feedback:
Create your own user feedback survey
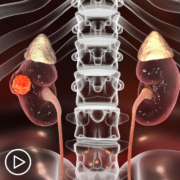
Biomarker CA-125 and Renal Medullary Carcinoma: What Do We Know?
Biomarker CA-125 and Renal Medullary Carcinoma: What Do We Know? from Patient Empowerment Network on Vimeo.
What is known about the renal medullary carcinoma (RMC) biomarker called CA-125? Expert Dr. Nizar Tannir explains how the CA-125 biomarker has been analyzed and further studies of the biomarker used along with other RMC testing.
Dr. Nizar Tannir is a Professor in the Department of Genitourinary Medical Oncology, Division of Cancer Medicine at The University of Texas MD Anderson Cancer Center.
[ACT]IVATION TIP
“…another valuable information to have, but it has to be interpreted in the context of other things in the context of how the patient is doing, whether they have symptoms that are improving or getting worse, and imaging studies. So all of this has to be incorporated, integrated together as one package and not just look at it individually and make decisions, right, only based on that. So it’s a good tool, it’s something to use, and we are learning more about it, and pretty soon, hopefully we will publish our results about that.”
Download Guide | Descargar Guía
Related Resources:
|

What Renal Medullary Carcinoma Treatment Options Are Available? |

Should Families of Renal Medullary Carcinoma Patients Undergo Genetic Testing? |
Transcript:
Cora:
Dr. Tannir, there is news about monitoring levels of the biomarker CA-125 and renal medullary carcinoma and how it may help predict disease development. What do we know about CA-125 as a biomarker in renal medullary carcinoma?
Dr. Tannir:
The CA-125 biomarker has been established as a good serological marker that you test in serum in women with ovarian cancer. So in women with ovarian cancer or suspected to have ovarian cancer, that blood test can be very valuable and elevated blood level of CA-125 in women with suspected ovarian cancer or established already women already diagnosed with ovarian cancer can provide information about the activity of the disease and response to therapy. So when a patient with ovarian cancer, for example, a response has, say, a debulking surgery and then the blood test is drawn, is tested after surgery the blood level will go down. You give them chemotherapy and the blood level goes down indicating that they’re responding. If they achieve a complete remission, complete response that CA-125 level could go down to undetectable level or normal range level as physiologic and women who don’t have ovarian cancer. Likewise, we use it in ovarian cancer to see if it’s rising, that the patient could be developing a recurrence of that disease. So it’s been very helpful in that regard.
And actually the pioneer this researcher, the physician scientist who led the initial work with CA-125 as a biomarker in cancer, was Dr. Robert Bast from MD Anderson. Now recently Dr. Msaouel and our team at MD Anderson looked at a battery, a panel of serology to really see if one of them is associated or could be linked to RMC and among a battery, a panel of several of these serological biomarkers. We found that CA-125 actually is a valuable blood test that we use to monitor RMC, not just in women like ovarian cancer, women with ovarian cancer, but in men and women with RMC the CA-125 could be valuable. Now, it’s not as they say panacea, it’s not like it’s if a patient has…the patient could have high tumor burden with obvious disease when you do CAT scans and MRIs and PET scans and other imaging studies.
But CA-125 may not be elevated but in many patients it can track the disease response to therapy that you are treating the patients with. And it could be valuable in that sense. But more recently we found that this could be also a relevant or a potentially relevant and important target to use therapy against RMC with that. So we are developing, Dr. Msaouel is preparing and hopefully we will launch this trial before the end of the year where we are using novel therapy based on that link based on that CA-125, whether this therapy will be more effective than chemotherapy, time will tell. So my activation tip on this is it’s good information to have, I encourage providers who treat patients with RMC at other institutions to test draw the blood order this test on their patients and see if in their patients it’ll be valuable.
It’ll be helpful to help them to track the disease. So that’s my activation tip is another valuable information to have, but it has to be interpreted in the context of other things in the context of how the patient is doing, whether they have symptoms that are improving or getting worse, and imaging studies. So all of this has to be incorporated, integrated together as one package and not just look at it individually and make decisions, right, only based on that. So it’s a good tool, it’s something to use, and we are learning more about it, and pretty soon, hopefully we will publish our results about that.
